{kind=link}

Designing correct all-atom protein constructions is a vital problem in bioengineering, because it entails producing each 3D structural information and 1D sequence data to outline side-chain atom placements. Most approaches at the moment rely closely on resolved experimentally decided structural datasets, that are scarce and biased, thereby limiting exploration of the pure protein area. Furthermore, these approaches usually compartmentalize the processes of sequence and construction technology, which ends up in inefficiencies and an inadequate integration of multimodal features. Addressing these obstacles is essential for progressing protein design, and facilitating developments in drug discovery, molecular engineering, and artificial biology.
Typical strategies of designing proteins depend on backbone-structure prediction or inverse-folding fashions to deduce sequences from recognized constructions. These methodologies are restricted by the separation of modalities, provided that quite a few methods deal with sequence and construction as distinct duties, thereby complicating their integration. A big dependence on experimentally decided structural datasets precludes a substantial variety of non-crystallizable proteins and restricts generalizability. The method of alternating between construction prediction and sequence technology prolongs runtime and heightens the danger of error propagation. Present instruments additionally face challenges in with the ability to match large-scale, function-specific protein designs as a result of inflexible constructions and limitations within the datasets. These limitations restrict the scope of current methodologies, significantly concerning the design of proteins that require excessive multimodal constancy and variation.
Researchers from UC Berkeley, Genentech, Microsoft Analysis, and New York College introduce PLAID (Protein Latent Induced Diffusion), a generative framework that synthesizes sequence and all-atom protein constructions by leveraging latent diffusion. Such a framework makes use of ESMFold-derived cohesive latent area for clean multimodal integration and reduces dependence on outdoors predictive fashions. This strategy will increase the information distribution by 2–4 orders of magnitude over structural datasets to cowl pure protein variety higher. It applies a Diffusion Transformer structure designed to be hardware-aware and scale up for conditional multimodal protein technology. Its operate and organism-conditioned samples attain excessive structural constancy and biophysical relevance throughout a large spectrum of protein design wants. These options describe huge strides ahead towards making numerous, scalable, and application-driven protein technology potential.
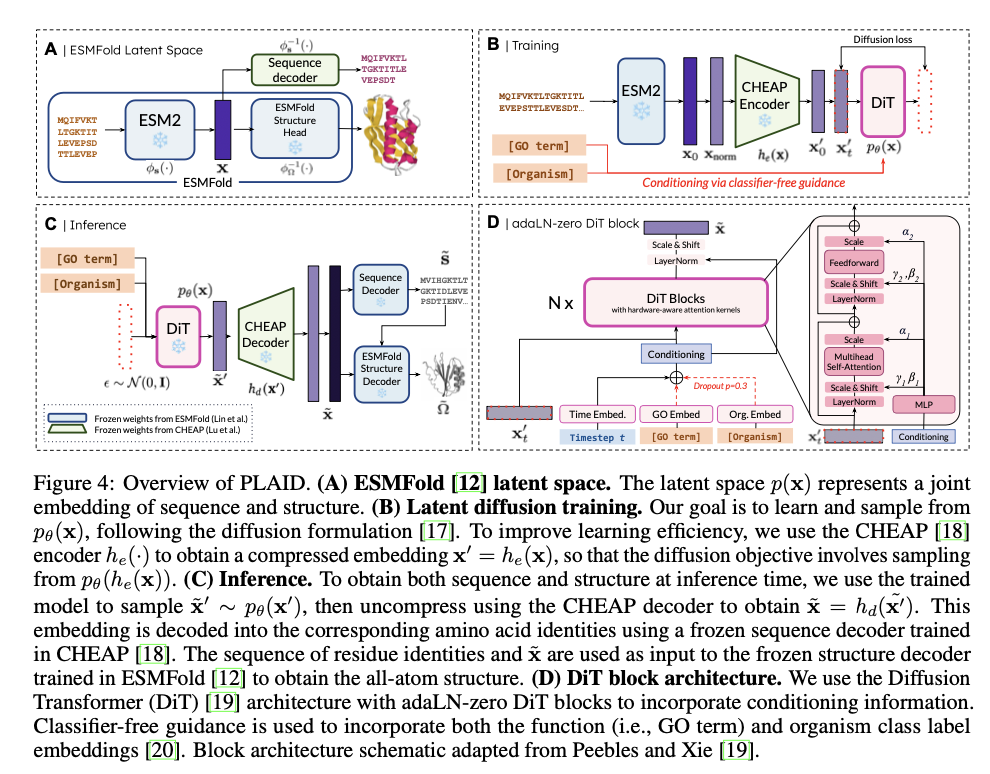
PLAID operates by correlating protein sequences to a decreased latent area, fine-tuned for environment friendly diffusion and technology. Oblique acquisition of structural constraints happens with using pre-trained ESMFold weights, owing to plentiful annotated sequence information in databases like Pfam. A discrete-time diffusion framework systematically enhances latent embeddings whereas integrating classifier-free steering for conditioning associated to operate and organism. It incorporates a CHEAP autoencoder for dimensionality discount and Diffusion Transformer blocks for denoising, thus making it scalable for lengthy protein sequences. Metrics embody cross-modal consistency, self-consistency, and conformity to pure protein biophysical distributions, validating the standard and organic relevance of generated samples.
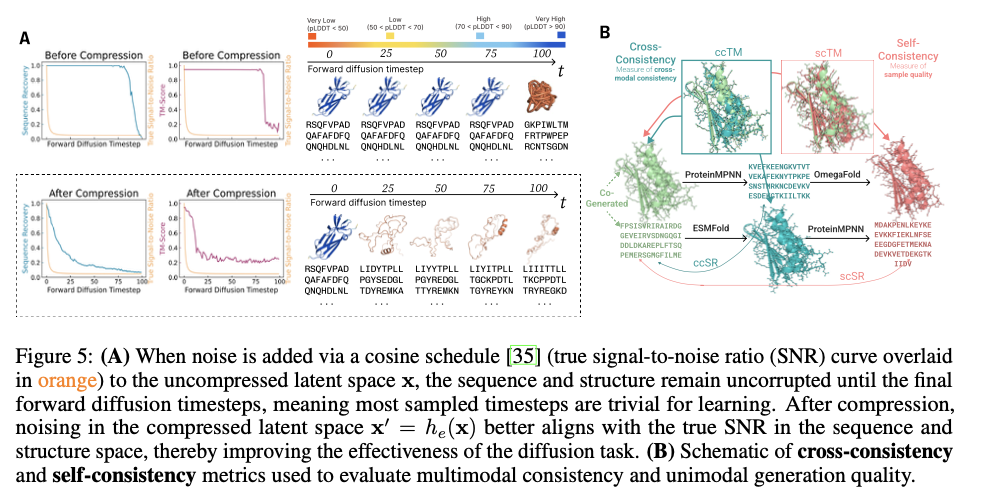
This progressive framework demonstrates substantial developments in producing biologically related and structurally correct protein designs. PLAID is extremely cross-modal constant and due to this fact demonstrates substantial enhancements within the alignment of generated sequences and constructions and likewise presents an improved designability relative to earlier approaches. The proteins produced display strong structural constancy, capturing the biophysical properties reminiscent of hydropathy, molecular weight, and variety in secondary construction, thus carefully resembling the pure proteins. Additionally, function-conditioned samples display elevated specificity, exactly reproducing motifs of lively websites and patterns of hydrophobicity, and with preserved sequence variety. When it comes to high quality, variety, and novelty metrics, PLAID demonstrates a constant superiority over baseline fashions, yielding the best amount of distinct and designable protein clusters that exhibit sensible structural and useful traits.
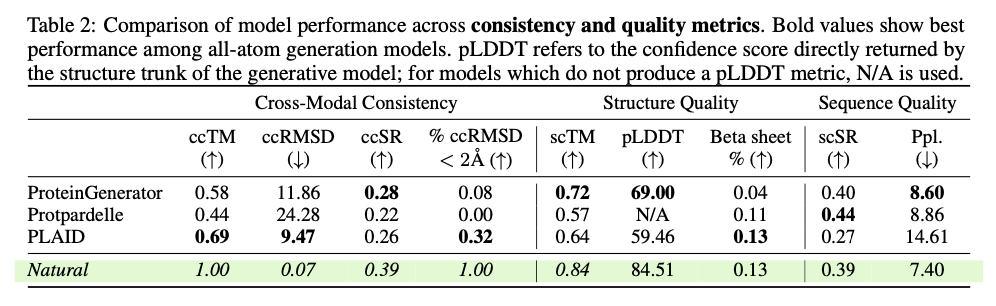
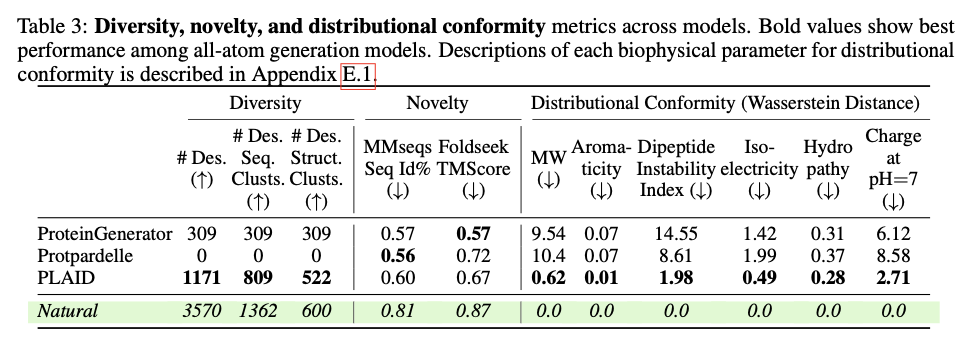
PLAID presents a revolutionary methodology for protein design by specializing in scalability, information availability, and multi-modality integration of main challenges. This fashion, it manages to hyperlink sequence and construction technology by making use of sequence-based coaching and latent diffusion for creating numerous, correct, and biologically related protein designs. This strategy can probably be used to additional purposes in molecular engineering, therapeutic improvement, and artificial biology and open up the doorways for brand new options for complicated organic issues.
Take a look at the Paper and GitHub Web page. All credit score for this analysis goes to the researchers of this challenge. Additionally, don’t neglect to observe us on Twitter and be part of our Telegram Channel and LinkedIn Group. Don’t Overlook to hitch our 60k+ ML SubReddit.
Aswin AK is a consulting intern at MarkTechPost. He’s pursuing his Twin Diploma on the Indian Institute of Expertise, Kharagpur. He’s obsessed with information science and machine studying, bringing a powerful tutorial background and hands-on expertise in fixing real-life cross-domain challenges.